




We use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies
As the core link of the gene expression regulation network, the interaction between RNA and protein plays a vital role in cell growth, differentiation, aging, and the occurrence and development of diseases. RNA Immunoprecipitation Sequencing (RIP-Seq) technology is a key tool to analyze RNA-protein interaction genomics, which skillfully combines RNA immunoprecipitation and high-throughput sequencing technology, and can systematically explore the dynamic interaction between them at the level of the complete transcriptome.
In recent years, with the iterative innovation of technology and the continuous expansion of application scenarios, RIP-Seq has made great achievements in disease mechanism analysis, new biomarker mining, and drug target screening. However, the complexity of technical principles, the meticulous requirements of experimental procedures, and the deep challenges of data analysis also mean that there are still many bottlenecks to be broken through in this field.
This paper comprehensively introduces RIP-Seq technology, including its principle, experimental process, data analysis steps, and expounds its application and value in gene expression regulation and disease research.
RIP-Seq technology combines RNA Immunoprecipitation (RIP) and high-throughput sequencing. RNA immune coprecipitation technology originated from protein immune coprecipitation (Co-IP). Its basic principle is to capture the RNA bound to the target protein in the cell lysate by using the specific binding of antigen and antibody, to study the interaction of RNA-protein complex in the natural state of cells.
High-throughput sequencing technology can quickly and efficiently determine a number of nucleic acid sequences. After the combination of the two, RIP-Seq can not only identify RNA interacting with specific proteins, but also perform RNA-Seq analysis on these RNAs in the whole transcriptome range, which greatly expands the depth and breadth of research.
The development of RIP-Seq technology is closely related to the demand for life science research and technical progress. In the early days, researchers used traditional RIP experiments combined with Northern Blot and RT-PCR to detect a small amount of known RNA, which was inefficient and limited in flux.
With the rapid development of high-throughput sequencing technology and the gradual integration of RIP and sequencing technology, RIP-Seq technology came into being. Nowadays, RIP-Seq has become a key technology to study RNA-protein interaction networks, which is widely used in many fields, such as gene expression regulation, disease marker screening, drug target discovery, and so on.
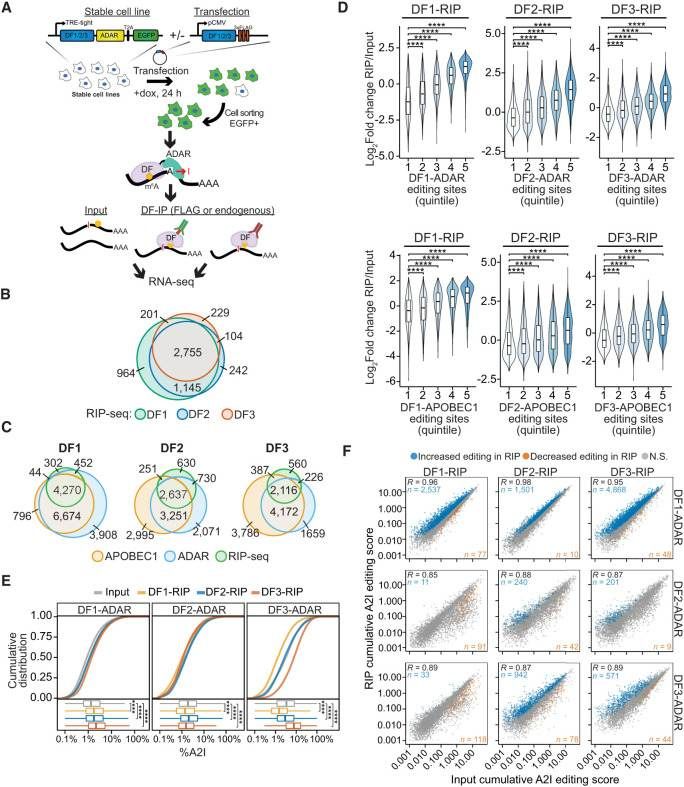 RIP-seq confirms cobinding of DF proteins to the same mRNA molecules (Flamand et al., 2022)
RIP-seq confirms cobinding of DF proteins to the same mRNA molecules (Flamand et al., 2022)
RIP borrows principles from protein immunoprecipitation (Co-IP) but focuses on RNA-protein complexes. The process begins with:
Cell Lysis: Gentle disruption of cells using detergents like Triton X-100, paired with protease and RNase inhibitors. Release complexes intact while preventing degradation. For sensitive cell types (e.g., primary neurons), optimized lysis buffers with reduced detergent concentrations may be necessary to avoid premature complex dissociation.
Antibody Selection: The backbone of RIP specificity. Monoclonal antibodies (mAbs) are prized for their high selectivity, minimizing non-specific binding—a critical factor in reducing background noise. Polyclonal antibodies, while more promiscuous, can capture multiple epitopes on the target protein, enhancing recovery rates for low-abundance interactors.
Immunoprecipitation: After antibody-incubation, Protein A/G beads (or magnetic beads for high-throughput workflows) capture the antibody-protein-RNA complexes. Stringent washing protocols follow, using buffers with varying salt concentrations (e.g., low-salt for weak interactors, high-salt for stringent purification) to remove unbound molecules without disrupting the complexes.
Once RNA is isolated from complexes, sequencing transforms raw molecules into biological insights:
RNA Fragmentation: Long RNA transcripts (e.g., mRNAs, lncRNAs) are sheared into 100–300 bp fragments. Chemical fragmentation (e.g., zinc-induced hydrolysis) offers random cleavage, while enzymatic methods (e.g., RNase III) produce more uniform sizes. The choice depends on downstream applications—random fragmentation is ideal for transcriptome-wide mapping, while enzymatic methods suit targeted analyses.
Library Construction: A multi-step process to prepare fragments for sequencing.
Sequencing Platforms: Illumina's NovaSeq and HiSeq dominate due to their accuracy (99.9% base call accuracy) and scalability. For rare cell populations or low-abundance RNAs, higher sequencing depths (e.g., 50–100 million reads per sample) are recommended to ensure statistical power.
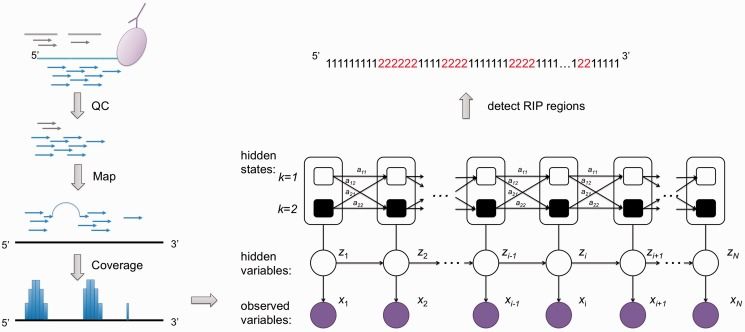 Simplified biological principles of RIP-seq (Li et al., 2013)
Simplified biological principles of RIP-seq (Li et al., 2013)
Service you may intersted in
Learn More:
RIP-Seq generates terabytes of data, requiring a robust analytical pipeline to transform raw sequences into biological meaning. Below is a step-by-step breakdown.
The original sequencing data often contains low-quality sequences, linker contamination, and sequencing errors, so it is necessary to control the quality of the data first.
After obtaining clean data, it is necessary to compare the sequencing reads to the reference genome or transcriptome.
Gene expression quantification of RIP-Seq data is an important step to analyze the RNA abundance bound to the target protein.
Differential expression analysis aims to find out the genes with significant differences in RNA expression binding to the target protein between the experimental group and the control group. Tools like DESeq2 or edgeR identify RNAs with significantly altered binding to the target protein between conditions (e.g., treated vs. untreated cells).
After screening differentially expressed RNA, it is very important to further explore its biological function.
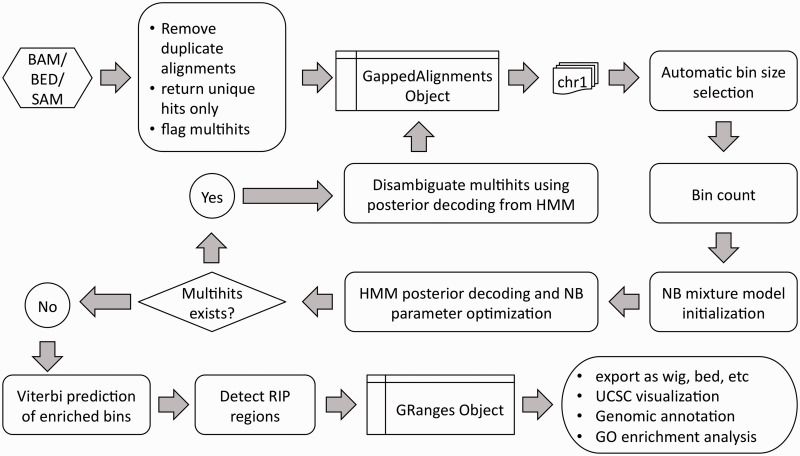 Detailed workflow of RIPSeeker (Flamand et al., 2022)
Detailed workflow of RIPSeeker (Flamand et al., 2022)
After completing the above analysis, various visualization tools (such as ggplot2, Circos, etc.) are used to display the analysis results in the form of charts, which can present the data characteristics and laws more intuitively.
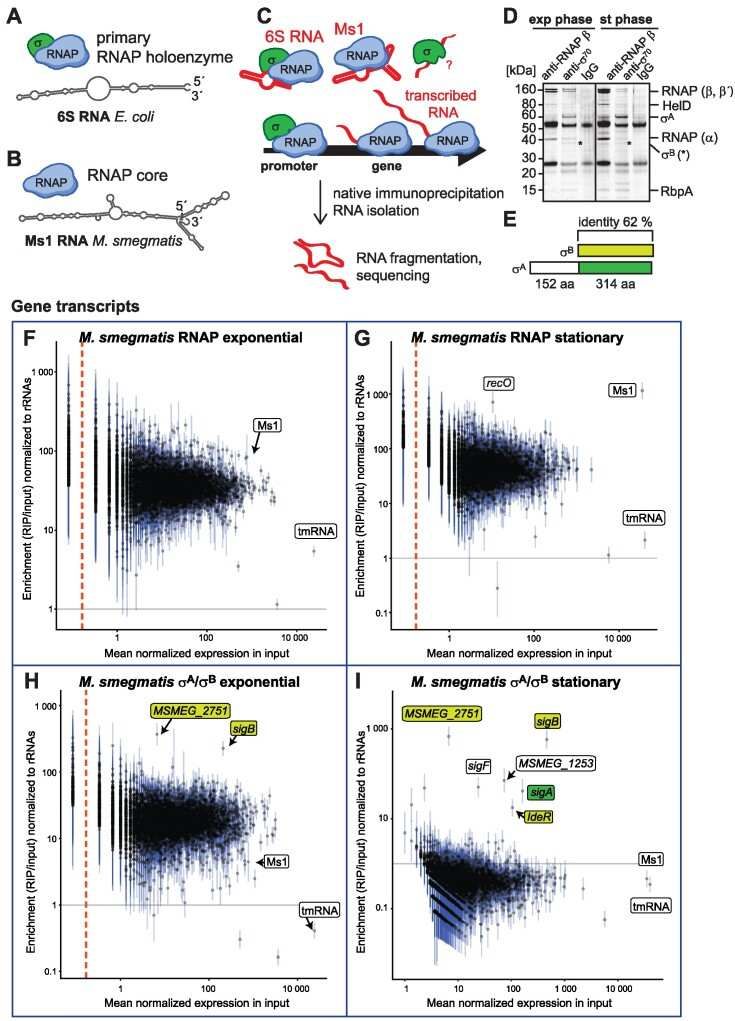 RIP-seq in Mycobacterium smegmatis (Vaňková et al., 2024)
RIP-seq in Mycobacterium smegmatis (Vaňková et al., 2024)
Service you may intersted in
In the RIP-Seq experiment, the key control experiment design is the core link to ensure the reliability of the results. It eliminates non-specific interference and verifies the validity of the experiment by setting a reasonable contrast, just like a "ruler" to measure the reliability of data.
IgG control is an important negative control in the RIP-Seq experiment. IgG is a kind of immunoglobulin and does not have the ability to specifically recognize the target protein.
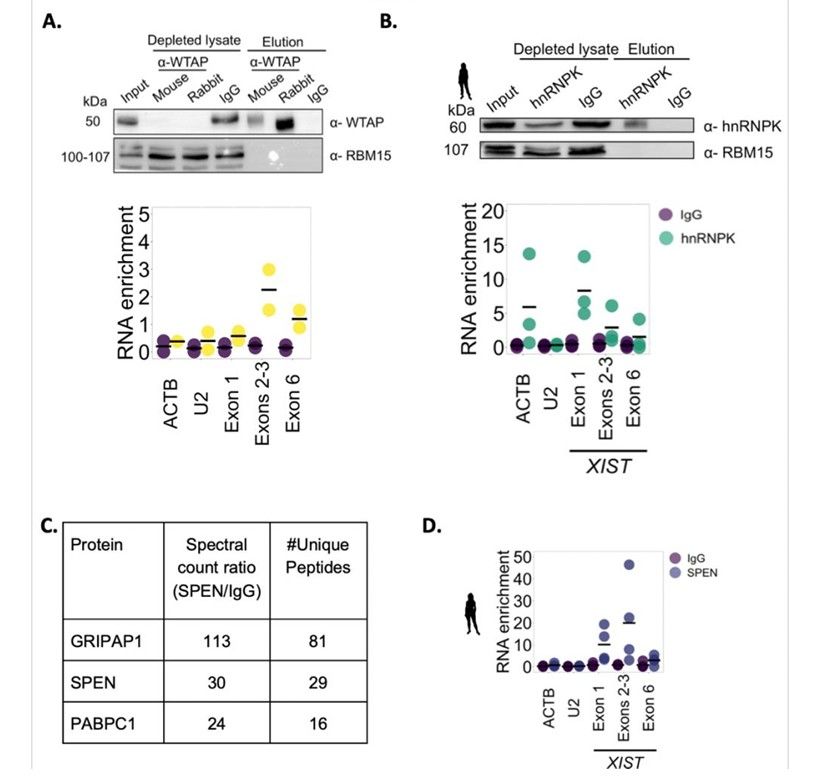 Human XIST interacts with WTAP, HNRNPK and SPEN (Ioannis et al., 2023)
Human XIST interacts with WTAP, HNRNPK and SPEN (Ioannis et al., 2023)
Input RNA control refers to RNA samples directly extracted from cell lysate before RNA immunoprecipitation. This sample represents the overall situation of all RNA in the cell and serves as a reference standard for the experiment. The library of Input RNA samples was constructed and sequenced, and compared with the sequencing results of the experimental group samples, the enrichment multiple of the target RNA in the immunoprecipitation process could be calculated. By analyzing the enrichment multiple, we can:
To sum up, RIP-Seq technology provides a powerful technical means for studying RNA-protein interaction by skillfully combining RNA immunoprecipitation with high-throughput sequencing. During the experiment, every step from cell lysis, antibody selection, to library sequencing, as well as the design of key control experiments such as IgG control and Input RNA control, all play a decisive role in the accuracy and reliability of the experimental results. With the continuous development and improvement of technology, RIP-Seq technology will play a more important role in the field of life science research and reveal more mysteries of life phenomena for us.
References
Terms & Conditions Privacy Policy Copyright © CD Genomics. All rights reserved.